Protein extraction
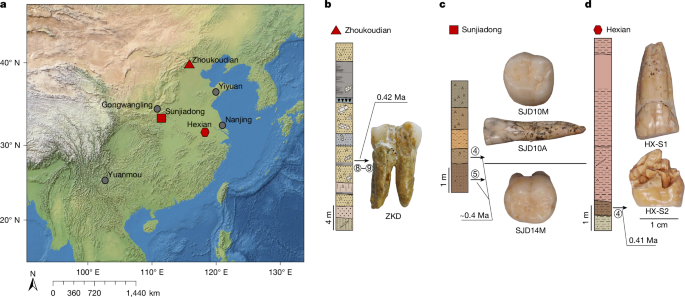
Animal fossils
Powder was drilled from the dentin (Supplementary Data 1), then mixed with 1.5 ml of 0.6 M HCl for decalcification. The precipitate was washed with ultrapure water at least 3 times until the pH reached approximately 7. Then, 200 µl of 50 mM NH4HCO3 was added, and the mixture was incubated at 65 °C for 3 h to extract soluble proteins. The supernatant containing the proteins was then transferred into a new centrifuge tube, and 1 µg of trypsin (Promega) was added. The mixture was incubated at 37 °C for 18 h. A 2.5% trifluoroacetic acid (TFA) solution (final concentration 0.1%) was added to stop the reaction. Subsequently, the trypsinized peptides were desalted and purified using C18 ZipTips57. For enamel samples, the powder was mixed with 1 ml of 5% HCl37 for decalcification, and the acid was replaced daily until the reaction ceased. The acid solution containing dissolved enamel peptides was concentrated using a vacuum concentrator, and the peptides were then desalted and purified using C18 ZipTips58.
Peptides were eluted into a solution of 80% acetonitrile with 0.1% TFA for MALDI-TOF mass spectrometry analysis. All sample preparations were performed in the dedicated clean room at the Molecular Paleontology Laboratory, IVPP of the Chinese Academy of Sciences in Beijing.
Hominin fossils
An acid etching method was used to extract protein from tooth enamel, modified from the process described in ref. 37. Disposable toothbrushes were used to remove surface contaminants from a small area of the enamel for etching. At the same time, the remaining teeth were wrapped with parafilm to prevent contact with any liquids. Before etching, the small enamel area was initially washed with 3% H2O2 for 30 s, followed by a rinse with ultrapure water. Approximately 100 µl of 5% (v/v) HCl was placed in the cap of a 1.5-ml microcentrifuge tube. A 2-min etch was performed by immersing the etching region in the HCl solution, and the initial etch solution was discarded. A second etch, lasting 15 min, was carried out in the cap of another separate microcentrifuge tube, and the etch solution was retained. This second etch was repeated, and the etch solutions were combined. After etching, the etched area was treated with 100 µl of 50 mM ammonium bicarbonate solution for 1 min to neutralize the acid. It was then rinsed with ultrapure water for 30 s and dried. The combined etch solution was then desalted using C18 ZipTips (Thermo Fisher Scientific) and eluted into a solution of 0.1% TFA and 80% acetonitrile (ACN). The peptide mixture (50 µl) was further divided into three aliquots; one aliquot was composed of 16 µl, among which 3 μl was used for the MALDI-TOF mass spectrometry test and 13 µl was retained as backup in our laboratory; two aliquots (each composed of 17 µl) were dried for LC–MS/MS analysis in two independent laboratories. All sample preparation for the experiment was conducted in the dedicated clean room at the Molecular Paleontology Laboratory, IVPP of the Chinese Academy of Sciences in Beijing.
MALDI-TOF mass spectrometry analysis
The peptide mixture was analysed on a Bruker autoflex maX MALDI-TOF mass spectrometer. In detail, 1 µl of peptide mixture was spotted onto a MTP384 Bruker ground-steel MALDI target plate, and 1 µl of α-cyano-4-hydroxycinnamic acid matrix solution (1% in 50% ACN/0.1% TFA (v/v/v)) was added on top. They were mixed, dried, and analysed on the mass spectrometer with a m/z range of 700–3,500. Each sample was analysed in triplicate. The raw data files were processed by mMass (v5.5.0)59.
LC–MS/MS analysis
For each hominin fossil, the eluted peptides from the enamel extraction were analysed under DDA mode in triplicate, including two runs on an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) at Capital Medical University, Beijing, and one run on an Orbitrap Exploris 480 mass spectrometer (Thermo Fisher Scientific) at Fudan University. Both devices were coupled to an Easy nLC 1200 HPLC system (Thermo Fisher Scientific).
For the Orbitrap Fusion Lumos at Capital Medical University, the peptides were initially loaded onto a 100 μm internal diameter × 2 cm trap column and then separated on a 150 μm internal diameter × 15 cm analytical column. Both columns were packed in-house using 3 μm reversed-phase silica (Reprosil-Pur C18 AQ, Dr. Maisch). The peptides were eluted using a 120 min linear gradient programme (0–8 min, 7–11% B; 8–96 min, 11–28% B; 96–108 min, 28–40% B; 108–113 min, 40–90% B; 113–120 min, 90% B) at a flow rate of 500 nl min−1. Buffer A was 0.1% formic acid in water, and buffer B was 80% acetonitrile and 0.1% formic acid. The MS1 data were acquired across 375–1,400 m/z, with a resolution of 120k at m/z 200, a 250% AGC target, and a maximum injection time of 50 ms. The MS2 scans were performed with a resolution of 15k at m/z 200, an AGC target of 100%, a 35% normalized collision energy, and a maximum injection time of 22 ms.
For the Orbitrap Exploris 480 at Fudan University, Shanghai, the peptides were separated on a 75 μm internal diameter × 25 cm analytical column, which was packed in-house using reversed-phase silica of 1.9 μm (Reprosil-Pur C18 AQ, Dr. Maisch). Buffer A was 0.1% formic acid in water, and buffer B was 80% acetonitrile and 0.1% formic acid. An 80 min gradient was used with the following profile: 5–8% B, 2 min, at a flow rate of 200 nl min−1; 8–44% B, 38 min, 200 nl min−1; 44–70% B, 8 min, 200 nl min−1; 70–100% B, 2 min, 200 nl min−1; 100% B, 10 min, 200 nl min−1; 100–5% B, 2 min, 200 nl min−1; 5% B, 2 min, 300 nl min−1; 5–100% B, 6 min, 300 nl min−1; 100% B, 10 min, 300 nl min−1. Full mass spectrometry scans were acquired for the first 65 min, after which the column was washed and re-equilibrated for 15 min without data acquisition. The full mass spectrometry data acquisition was conducted across the range of m/z 350–1,600, with a resolution of 60k at m/z 200. The AGC target was set to ‘standard’, and the maximum injection time mode was set to ‘auto’. The MS/MS spectra were acquired with a resolution of 15k at m/z 200, a maximum injection time of 30 ms and a normalized collision energy of 30%. The AGC target was also set to standard.
For each animal fossil, the eluted peptides from enamel extractions were analysed for one run under DDA mode on the Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific), either at Capital Medical University or Fudan University. For the Orbitrap Fusion Lumos at Capital Medical University, the liquid chromatography gradient and mass spectrometry parameters were the same as those of the hominin fossils. The Orbitrap Fusion Lumos at Fudan University was also interfaced with an Easy nLC 1200 HPLC system (Thermo Scientific). The peptides were separated on a 75 μm internal diameter × 20 cm analytical column packed with 1.9 μm reversed-phase silica. Mobile phase A consisted of 0.1% formic acid, and mobile phase B consisted of 80% acetonitrile and 0.1% formic acid. An 80 min gradient was used with the following profile: 2–5% B, 3 min, at a flow rate of 200 nl min−1; 5–35% B, 40 min, 200 nl min−1; 35–44% B, 5 min, 200 nl min−1; 44–100% B, 2 min, 200 nl min−1; 100% B, 10 min, 200 nl min−1; 100–5% B, 2 min, 200 nl min−1; 5% B, 2 min, 300 nl min−1; 5–100% B, 6 min, 300 nl min−1; 100% B, 10 min, 300 nl min−1. Full mass spectrometry scans were acquired for the first 65 min, after which the column was washed and re-equilibrated for 15 min without data acquisition. The full mass spectrometry data acquisition was conducted across the m/z range 350–1,600, with a resolution of 60k at m/z 200, a 100% AGC target, and a maximum injection time of 50 ms. The MS/MS spectra were acquired with a resolution of 15k at m/z 200, a 100% AGC target, a 30% normalized collision energy, and a maximum injection time of 30 ms. Blank extractions were processed concurrently to monitor the exogenous contaminants during the procedure.
Data search strategy
MaxQuant (v2.6.0.0)32, PEAKS Online (v12)33 and pFind (v3.2.1)34 were used to search the raw data. The H. erectus raw files were searched with the corresponding laboratory blanks and modern H. sapiens raw files43 against the ‘Hominidae enamel database’, supplemented with the contaminant database. Unspecific digestion was selected in each software. The animal raw files were searched against the ‘mammal enamel database’.
Database composition
The ‘Hominidae enamel database’ comprised 13 selected enamel proteins from Hominidae. Besides the commonly used 12 proteins (AHSG, ALB, AMBN, AMELX, AMELY, AMTN, COL17A1, ENAM, KLK4, MMP20, ODAM and TUFT1), SERPINC1 was also added because we identified this protein in Harbin with abundant peptides and elevated deamidation rates (nearly 100%), and this protein was also reported in modern enamel60,61. The sequences were retrieved from UniProt, downloaded from the ‘Hominid Palaeoproteomic Reference Dataset’ (https://zenodo.org/records/7728060), translated from the genomes of public projects30, and specific sequences from published palaeoproteomes11. The ‘mammal enamel database’ was composed of mammal sequences of the same enamel proteins above, retrieved from UniProt using the gene names and ‘Mammalia (mammals) [40674]’. The contaminant database was composed of a previously published contaminant database62, the cRAP database (https://www.thegpm.org/crap/), and the contaminant database from MaxQuant (v2.6.0.0). The ‘Hominidae enamel database’ was accessible through the ProteomeXchange Consortium (Data availability).
PEAKS search
The precursor ion (MS1) mass tolerance was set to 10 ppm, with a fragment ion (MS2) mass tolerance of 0.02 Da for all PEAKS searches, with unspecific digestion. Variable modifications included deamidation (NQ), oxidation (M), hydroxylation (P), phosphorylation (STY), N-terminal pyro-Glu from E, and N-terminal pyro-Glu from Q, with no fixed modifications and up to three modifications allowed per peptide. The peptide length was set to 6–45. PSMs were filtered using a false discovery rate (FDR) of 1%, and proteins were filtered with criteria of −10logP ≥ 20 and average local confidence (ALC) ≥ 50% (de novo only).
After our initial search with PEAKS, additional variable modifications were included in the second-round search for Hexian H. erectus samples: chlorination and dichlorination of tyrosine residues, dehydration, dioxidation (W), carbonyl E, dioxidation (M), oxidation (HW), ornithine derived from arginine, tryptophan oxidation to kynurenine, tryptophan oxidation to oxolactone, and proline oxidation to pyroglutamic acid. Up to five modifications per peptide were permitted. The peptide length range was set to 6–30.
After removing peptides from the contaminant database and those detected in the extraction blank samples or matching multiple genes, the deamidation rates of glutamine (Q) and asparagine (N) were calculated for each sample based on PSM counts. We used auxiliary tools for PSM prediction with PEAKS.
pFind search
We included Deamidation [N], Deamidation [Q], Oxidation [M], Oxidation [P], Oxidation [W], Gln->pyro-Glu[AnyN-termQ], Glu->pyro-Glu[AnyN-termE], Pro->pyro-Glu[P], Phospho[S], Phospho[T], Phospho[Y], Dehydrated[S], Dehydrated[T], Dehydrated[Y], Arg->Orn[R], Dioxidation[M], Dioxidation[W], Thiazolidine[W], Trp->Kynurenin[W], Trp->Oxolactone[W], Ammonia-loss[N], His->Asp[H], Pro->HAVA[P], and Amidated[AnyC-term] as variable modifications, with no fixed modifications included. Spectra FDR was set at 1%, and protein FDR was 10%. The mass range of each peptide was set from 350 to 4,000 Da. Open search was enabled. Within pFind’s modification configuration, the default mass ‘X’ is preset to that of isoleucine/leucine. To avoid incorrect identification of the X residue, we set its mass to 6,228.71 Da (Sm41), which substantially exceeds the mass of any natural amino acid. Therefore, any in silico peptide with an X produces abnormal theoretical precursor and fragment ion masses, effectively preventing its matching to experimental spectra during database searches and thus reducing false positives from these ambiguous sequence regions. The other parameters were the same as the settings in PEAKS.
After our first-round search with pFind, some additional variable modifications were selected for inclusion in the second-round search for Hexian H. erectus samples: Chlorination[Y], dichlorination[Y], Carbonyl[E], and Dioxidation[P](Pro->Glu[P]).
MaxQuant search
No fixed modifications were specified. Variable modifications included Deamidation (NQ), Phosphorylation (STY), Gln to N-terminal pyro-Glu, Glu to N-terminal pyro-Glu, Dioxidation (MW), Oxidation (M), Oxidation (P), and Oxidation (W). PSM FDR was set at 1% for all 7 samples. The search also enabled the identification of dependent and secondary peptides. The remaining parameters were set to their default values. After the search, the deamidation rates of N and Q were calculated63 for each sample.
In addition, we determined the extra variable modification for Hexian H. erectus samples with PEAKS and pFind results. Cl(Y) and diCl (Y) were added for HX-S1 (both post-translational modifications were self-made in Configuration-Modifications), and Cl (Y) and Dehydrated (STY) were included for HX-S2.
Considering the high resolution of the instrumentation used, we also reduced the precursor ion (MS1) mass tolerance to 5 ppm and repeated all analyses in PEAKS and pFind. The results were similar, and the two main SAPs (AMBN 253 and AMBN 273) within the Homo genus were consistently identified (Supplementary Table 8). As the final results do not change significantly, we focus mainly on the results from the commonly used 10 ppm search to make our results more comparable to previous studies.
Construction of the consensus protein sequences and phylogenetic analysis
Consensus sequences of endogenous proteins were reconstructed for phylogenetic analysis (Supplementary Data 1). Peptides shorter than eight amino acids or with abnormal or artificial post-translational modifications were excluded for the consensus sequence reconstruction of each endogenous protein. Only alleles with a PSM count ≥2, a PSM ratio ≥10%, and an intensity ratio ≥10% were considered reliable and retained. For heterozygous sites, at least two peptides were required per allele. The heterozygous variant site, observed only in the Harbin specimen, was identified with 79 peptides for the V allele and 38 for the M allele at position 273 in AMBN (Table 1). We include both alleles of the Harbin consensus in Supplementary Data 2. We performed additional AMELY sequence correction as described in Supplementary Note 4.
The protein data in the phylogenetic tree include Denisova 3 (ref. 52), two Neanderthals (Altai, Vindija33.19)45,53, two modern humans from the HGDP project (San, HGDP0987; Bougainville, HGDP01027)30, a chimpanzee (Pan)29, and a Gorilla29. We used PartitionFinder2 (v2.1.1)64 to identify the best partitioning schemes and amino acid substitution models for our dataset. Then, we built a consensus Bayesian phylogenetic tree using the software MrBayes (v3.2.6)65, running 8 Markov Chain Monte Carlo (MCMC) chains for 1 million iterations in 2 independent runs. Sampling was done every 500 generations, and the first 200,000 iterations were discarded as burn-in. The tree was plotted with FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree).
DNA analysis
Sliding window analysis
Variant sites were identified where either Denisova 3 or two Neanderthals (Vindija33.19 and Altai) differed from two modern African diploid genomes (S_Khomani_San-1.DG, S_Mandenka-2.DG) from the phased Simons Genome Diversity Project panel (SGDP) (https://sharehost.hms.harvard.edu/genetics/reich_lab/sgdp/phased_data2021)66,67. For each of 129 overlapping 20 kb windows (with 2 kb steps), pairwise matching rates were calculated for variants between each African haploid and each archaic artificial haploid of either Denisova 3 or the Altai Neanderthal (where each variant is randomly assigned to a haploid to preserve all variants), covering in total 276 kb of DNA sequence surrounding the rs564905233 SNP. For each window, statistical significance of the differences between the African–Denisovan and African–Neanderthal matching rates were assessed using a Wilcoxon rank-sum test, and a permutation test (n = 1,000 permutations). The Wilcoxon rank-sum test was used to compare distributions of pairwise matching rates, with W representing the sum of ranks in the African–Denisovan group. The permutation test was used to evaluate differences in mean matching rates. No adjustments were made for multiple comparisons, since each window was analysed independently to identify localized regions of divergence. All statistical tests were performed as two-sided tests. Full results, including W, exact P values, and the number of comparisons per window, are provided in Supplementary Data 3.
Ethics statement
Permission to test for ancient proteins in the human specimens from this study was granted by the collection room of the IVPP, the Hexian Culture, Tourism, and Sports Bureau, and the Luanchuan County Culture, Radio, Television, and Tourism Bureau. The work was conducted in collaboration with local researchers, who are co-authors because of their contributions to assembling archaeological materials and/or discussions that informed the study.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.